3d_molecular_dynamics_output.pdb
AMBER
gromacs_1ns_trajectory.xtc
MM-PBSA
Complete molecular dynamics simulation from topology generation to MM-PBSA binding energy calculation. Three force fields, automated trajectory analysis, and interactive 3D visualization with PyMOLinteractive 3D.
Real molecular dynamics simulation output — AMBER99SB-ILDN force field, TIP3P solvent, 300K. This is actual computational output from our pipeline.
From raw PDB structure to binding energy calculation. Every stage is automated, validated, and reproducible.
Choose the force field best suited to your system. Each has been extensively validated for different biomolecular classes.
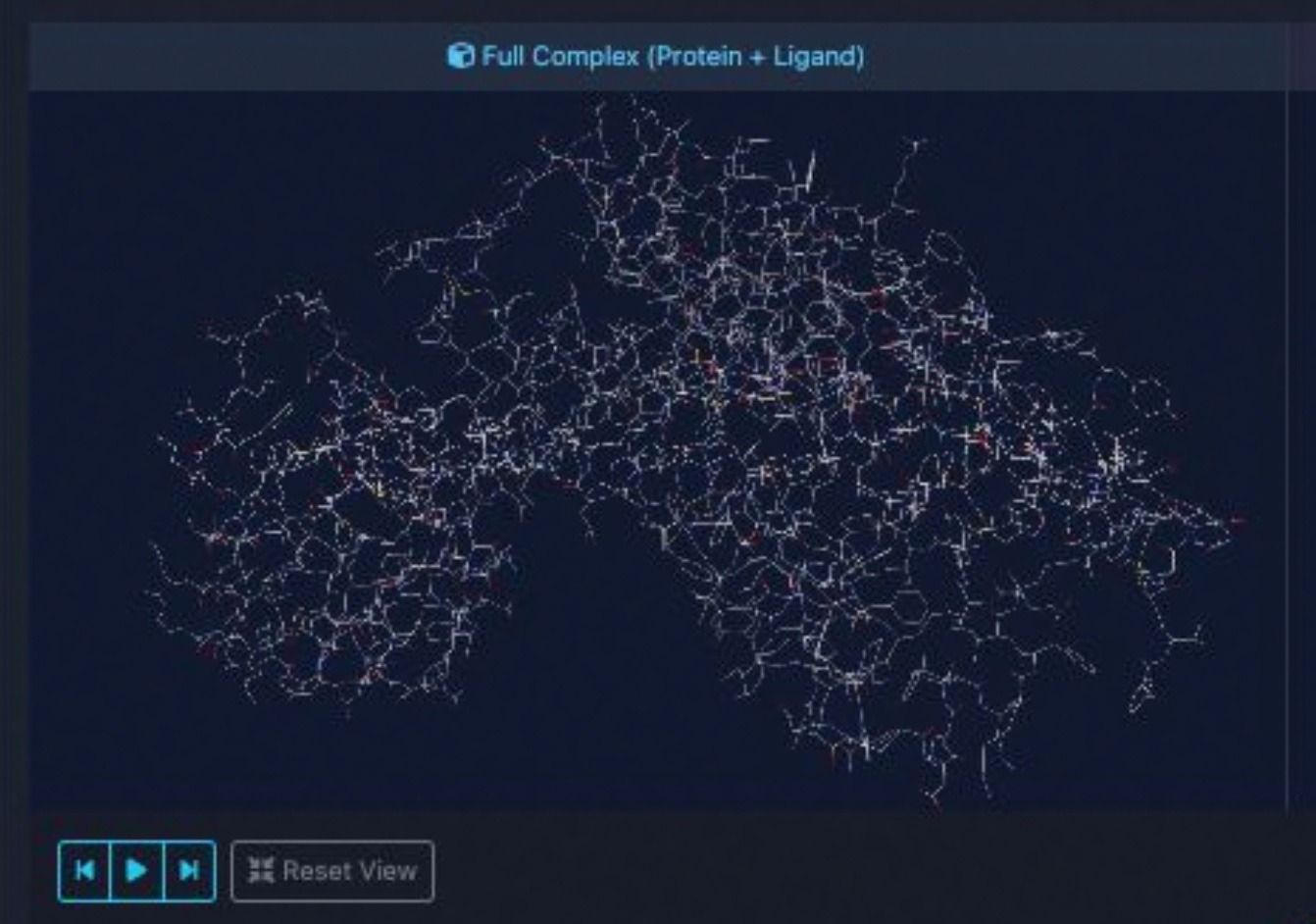
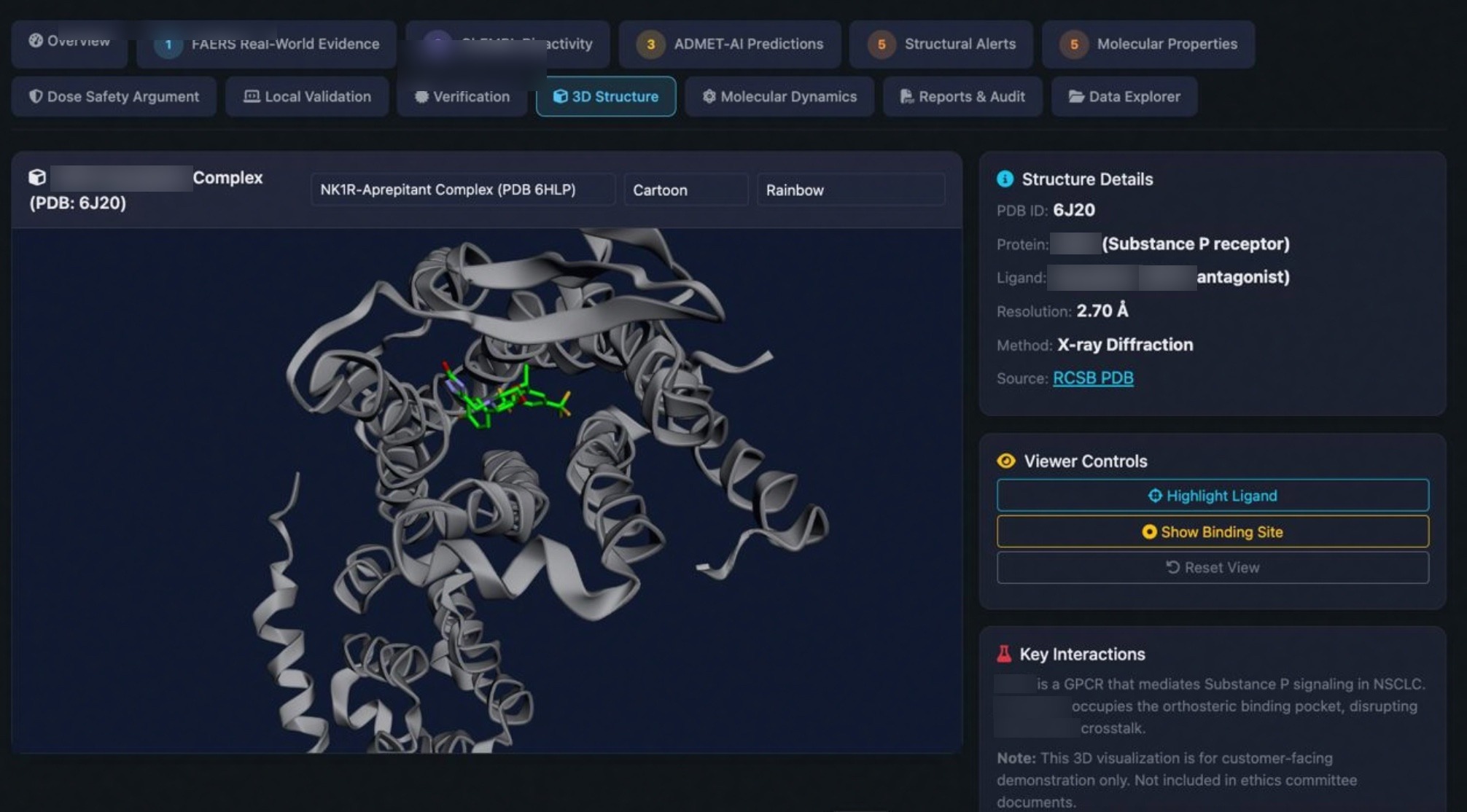
Every MD simulation includes interactive PyMOLinteractive 3D visualizations showing hydrogen bonds, binding hotspots, and real-time trajectory playback.
Quantitative binding energy calculation with per-residue decomposition. Identify the key interactions driving ligand binding.
The MM-PBSA method decomposes the binding free energy into physically meaningful components, revealing whether binding is driven by electrostatics, van der Waals contacts, or desolvation.
Submit your protein-ligand complex for a full molecular dynamics simulation with binding energy analysis and interactive 3D visualization.